Homologous recombination

Figure 1. During meiosis, homologous recombination can produce new combinations of genes as shown here between similar but not identical copies of human chromosome 1.
Homologous recombination is a type of genetic recombination in which nucleotide sequences are exchanged between two similar or identical molecules of DNA. It is most widely used by cells to accurately repair harmful breaks that occur on both strands of DNA, known as double-strand breaks (DSB). Homologous recombination also produces new combinations of DNA sequences during meiosis, the process by which eukaryotes make gamete cells, like sperm and egg cells in animals. These new combinations of DNA represent genetic variation in offspring, which in turn enables populations to adapt during the course of evolution.[1] Homologous recombination is also used in horizontal gene transfer to exchange genetic material between different strains and species of bacteria and viruses.
Although homologous recombination varies widely among different organisms and cell types, most forms involve the same basic steps. After a double-strand break occurs, sections of DNA around the 5' ends of the break are cut away in a process called resection. In the strand invasion step that follows, an overhanging 3' end of the broken DNA molecule then "invades" a similar or identical DNA molecule that is not broken. After strand invasion, the further sequence of events may follow either of two main pathways discussed below (see Models); the DSBR (double-strand break repair) pathway or the SDSA (synthesis-dependent strand annealing) pathway. Homologous recombination that occurs during DNA repair tends to result in non-crossover products, in effect restoring the damaged DNA molecule as it existed before the double-strand break.
Homologous recombination is conserved across all three domains of life as well as viruses, suggesting that it is a nearly universal biological mechanism. The discovery of genes for homologous recombination in protists—a diverse group of eukaryotic microorganisms—has been interpreted as evidence that meiosis emerged early in the evolution of eukaryotes. Since their dysfunction has been strongly associated with increased susceptibility to several types of cancer, the proteins that facilitate homologous recombination are topics of active research. Homologous recombination is also used in gene targeting, a technique for introducing genetic changes into target organisms. For their development of this technique, Mario Capecchi, Martin Evans and Oliver Smithies were awarded the 2007 Nobel Prize for Physiology or Medicine; Capecchi[2] and Smithies[3] independently discovered applications to mouse embryonic stem cells, however the highly conserved mechanisms underlying the DSB repair model, including uniform homologous integration of transformed DNA (gene therapy), were first shown in plasmid experiments by Orr-Weaver, Szostack and Rothstein.[4][5][6] Researching the plasmid-induced DSB, using γ-irradiation[7] in the 1970s-1980s, led to later experiments using endonucleases (e.g. I-SceI) to cut chromosomes for genetic engineering of mammalian cells, where nonhomologous recombination is more frequent than in yeast.[8]
Contents
1 History and discovery
2 In eukaryotes
2.1 Timing within the mitotic cell cycle
2.2 Preliminary steps
2.3 Models
2.3.1 DSBR pathway
2.3.2 SDSA pathway
2.3.3 SSA pathway
2.3.4 BIR pathway
3 In bacteria
3.1 RecBCD pathway
3.2 RecF pathway
3.3 Branch migration
3.4 Resolution
3.5 Facilitating genetic transfer
3.6 Bacterial transformation
4 In viruses
5 Effects of dysfunction
6 Evolutionary conservation
6.1 The RecA recombinase family
6.2 Meiosis-specific proteins
7 Technological applications
7.1 Gene targeting
7.2 Protein engineering
7.3 Cancer therapy
8 See also
9 References
10 External links
History and discovery
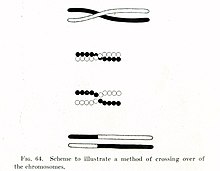
Figure 2. An early illustration of crossing over from Thomas Hunt Morgan
In the early 1900s, William Bateson and Reginald Punnett found an exception to one of the principles of inheritance originally described by Gregor Mendel in the 1860s. In contrast to Mendel's notion that traits are independently assorted when passed from parent to child—for example that a cat's hair color and its tail length are inherited independent of each other—Bateson and Punnett showed that certain genes associated with physical traits can be inherited together, or genetically linked.[9][10] In 1911, after observing that linked traits could on occasion be inherited separately, Thomas Hunt Morgan suggested that "crossovers" can occur between linked genes,[11] where one of the linked genes physically crosses over to a different chromosome. Two decades later, Barbara McClintock and Harriet Creighton demonstrated that chromosomal crossover occurs during meiosis,[12][13] the process of cell division by which sperm and egg cells are made. Within the same year as McClintock's discovery, Curt Stern showed that crossing over—later called "recombination"—could also occur in somatic cells like white blood cells and skin cells that divide through mitosis.[12][14]
In 1947, the microbiologist Joshua Lederberg showed that bacteria—which had been assumed to reproduce only asexually through binary fission—are capable of genetic recombination, which is more similar to sexual reproduction. This work established E. coli as a model organism in genetics,[15] and helped Lederberg win the 1958 Nobel Prize in Physiology or Medicine.[16] Building on studies in fungi, in 1964 Robin Holliday proposed a model for recombination in meiosis which introduced key details of how the process can work, including the exchange of material between chromosomes through Holliday junctions.[17] In 1983, Jack Szostak and colleagues presented a model now known as the DSBR pathway, which accounted for observations not explained by the Holliday model.[17][6] During the next decade, experiments in Drosophila, budding yeast and mammalian cells led to the emergence of other models of homologous recombination, called SDSA pathways, which do not always rely on Holliday junctions.[17]
Much of the later work identifying proteins involved in the process and determining their mechanisms has been performed by a number of individuals including James Haber, Patrick Sung, Stephen Kowalczykowski, and others.
In eukaryotes
Homologous recombination (HR) is essential to cell division in eukaryotes like plants, animals, fungi and protists. In cells that divide through mitosis, homologous recombination repairs double-strand breaks in DNA caused by ionizing radiation or DNA-damaging chemicals.[18] Left unrepaired, these double-strand breaks can cause large-scale rearrangement of chromosomes in somatic cells,[19] which can in turn lead to cancer.[20]
In addition to repairing DNA, homologous recombination also helps produce genetic diversity when cells divide in meiosis to become specialized gamete cells—sperm or egg cells in animals, pollen or ovules in plants, and spores in fungi. It does so by facilitating chromosomal crossover, in which regions of similar but not identical DNA are exchanged between homologous chromosomes.[21][22] This creates new, possibly beneficial combinations of genes, which can give offspring an evolutionary advantage.[23] Chromosomal crossover often begins when a protein called Spo11 makes a targeted double-strand break in DNA.[24] These sites are non-randomly located on the chromosomes; usually in intergenic promoter regions and preferentially in GC-rich domains[25] These double-strand break sites often occur at recombination hotspots, regions in chromosomes that are about 1,000–2,000 base pairs in length and have high rates of recombination. The absence of a recombination hotspot between two genes on the same chromosome often means that those genes will be inherited by future generations in equal proportion. This represents linkage between the two genes greater than would be expected from genes that independently assort during meiosis.[26]
Timing within the mitotic cell cycle
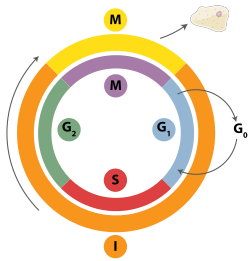
Figure 3. Homologous recombination repairs DNA before the cell enters mitosis (M phase). It occurs only during and shortly after DNA replication, during the S and G2phases of the cell cycle.
Double-strand breaks can be repaired through homologous recombination or through non-homologous end joining (NHEJ). NHEJ is a DNA repair mechanism which, unlike homologous recombination, does not require a long homologous sequence to guide repair. Whether homologous recombination or NHEJ is used to repair double-strand breaks is largely determined by the phase of cell cycle. Homologous recombination repairs DNA before the cell enters mitosis (M phase). It occurs during and shortly after DNA replication, in the S and G2 phases of the cell cycle, when sister chromatids are more easily available.[27] Compared to homologous chromosomes, which are similar to another chromosome but often have different alleles, sister chromatids are an ideal template for homologous recombination because they are an identical copy of a given chromosome. In contrast to homologous recombination, NHEJ is predominant in the G1 phase of the cell cycle, when the cell is growing but not yet ready to divide. It occurs less frequently after the G1 phase, but maintains at least some activity throughout the cell cycle. The mechanisms that regulate homologous recombination and NHEJ throughout the cell cycle vary widely between species.[28]
Cyclin-dependent kinases (CDKs), which modify the activity of other proteins by adding phosphate groups to (that is, phosphorylating) them, are important regulators of homologous recombination in eukaryotes.[28] When DNA replication begins in budding yeast, the cyclin-dependent kinase Cdc28 begins homologous recombination by phosphorylating the Sae2 protein.[29] After being so activated by the addition of a phosphate, Sae2 uses its endonuclease activity to make a clean cut near a double-strand break in DNA. This allows a three-part protein known as the MRX complex to bind to DNA, and begins a series of protein-driven reactions that exchange material between two DNA molecules.[30]
Preliminary steps
The packaging of eukaryotic DNA into chromatin presents a barrier to all DNA-based processes that require recruitment of enzymes to their sites of action. To allow HR DNA repair, the chromatin must be remodeled. In eukaryotes, ATP dependent chromatin remodeling complexes and histone-modifying enzymes are two predominant factors employed to accomplish this remodeling process.[31]
Chromatin relaxation occurs rapidly at the site of a DNA damage.[32] In one of the earliest steps, the stress-activated protein kinase, c-Jun N-terminal kinase (JNK), phosphorylates SIRT6 on serine 10 in response to double-strand breaks or other DNA damage.[33] This post-translational modification facilitates the mobilization of SIRT6 to DNA damage sites, and is required for efficient recruitment of poly (ADP-ribose) polymerase 1 (PARP1) to DNA break sites and for efficient repair of DSBs.[33]PARP1 protein starts to appear at DNA damage sites in less than a second, with half maximum accumulation within 1.6 seconds after the damage occurs.[34] Next the chromatin remodeler Alc1 quickly attaches to the product of PARP1 action, a poly-ADP ribose chain, and Alc1 completes arrival at the DNA damage within 10 seconds of the occurrence of the damage.[32] About half of the maximum chromatin relaxation, presumably due to action of Alc1, occurs by 10 seconds.[32] This then allows recruitment of the DNA repair enzyme MRE11, to initiate DNA repair, within 13 seconds.[34]
γH2AX, the phosphorylated form of H2AX is also involved in the early steps leading to chromatin decondensation after DNA double-strand breaks. The histone variant H2AX constitutes about 10% of the H2A histones in human chromatin.[35] γH2AX (H2AX phosphorylated on serine 139) can be detected as soon as 20 seconds after irradiation of cells (with DNA double-strand break formation), and half maximum accumulation of γH2AX occurs in one minute.[35] The extent of chromatin with phosphorylated γH2AX is about two million base pairs at the site of a DNA double-strand break.[35] γH2AX does not, itself, cause chromatin decondensation, but within 30 seconds of irradiation, RNF8 protein can be detected in association with γH2AX.[36] RNF8 mediates extensive chromatin decondensation, through its subsequent interaction with CHD4,[37] a component of the nucleosome remodeling and deacetylase complex NuRD.
After undergoing relaxation subsequent to DNA damage, followed by DNA repair, chromatin recovers to a compaction state close to its pre-damage level after about 20 min.[32]
Models
Two primary models for how homologous recombination repairs double-strand breaks in DNA are the double-strand break repair (DSBR) pathway (sometimes called the double Holliday junction model) and the synthesis-dependent strand annealing (SDSA) pathway.[38] The two pathways are similar in their first several steps. After a double-strand break occurs, the MRX complex (MRN complex in humans) binds to DNA on either side of the break. Next a resection, in which DNA around the 5' ends of the break is cut back, is carried out in two distinct steps. In the first step of resection, the MRX complex recruits the Sae2 protein. The two proteins then trim back the 5' ends on either side of the break to create short 3' overhangs of single-strand DNA. In the second step, 5'→3' resection is continued by the Sgs1 helicase and the Exo1 and Dna2 nucleases. As a helicase, Sgs1 "unzips" the double-strand DNA, while Exo1 and Dna2's nuclease activity allows them to cut the single-stranded DNA produced by Sgs1.[29]

Figure 4. The DSBR and SDSA pathways follow the same initial steps, but diverge thereafter. The DSBR pathway most often results in chromosomal crossover (bottom left), while SDSA always ends with non-crossover products (bottom right).
The RPA protein, which has high affinity for single-stranded DNA, then binds the 3' overhangs.[39] With the help of several other proteins that mediate the process, the Rad51 protein (and Dmc1, in meiosis) then forms a filament of nucleic acid and protein on the single strand of DNA coated with RPA. This nucleoprotein filament then begins searching for DNA sequences similar to that of the 3' overhang. After finding such a sequence, the single-stranded nucleoprotein filament moves into (invades) the similar or identical recipient DNA duplex in a process called strand invasion. In cells that divide through mitosis, the recipient DNA duplex is generally a sister chromatid, which is identical to the damaged DNA molecule and provides a template for repair. In meiosis, however, the recipient DNA tends to be from a similar but not necessarily identical homologous chromosome.[38] A displacement loop (D-loop) is formed during strand invasion between the invading 3' overhang strand and the homologous chromosome. After strand invasion, a DNA polymerase extends the end of the invading 3' strand by synthesizing new DNA. This changes the D-loop to a cross-shaped structure known as a Holliday junction. Following this, more DNA synthesis occurs on the invading strand (i.e., one of the original 3' overhangs), effectively restoring the strand on the homologous chromosome that was displaced during strand invasion.[38]
DSBR pathway
After the stages of resection, strand invasion and DNA synthesis, the DSBR and SDSA pathways become distinct.[38] The DSBR pathway is unique in that the second 3' overhang (which was not involved in strand invasion) also forms a Holliday junction with the homologous chromosome. The double Holliday junctions are then converted into recombination products by nicking endonucleases, a type of restriction endonuclease which cuts only one DNA strand. The DSBR pathway commonly results in crossover, though it can sometimes result in non-crossover products; the ability of a broken DNA molecule to collect sequences from separated donor loci was shown in mitotic budding yeast using plasmids or endonuclease induction of chromosomal events.[40][41] Because of this tendency for chromosomal crossover, the DSBR pathway is a likely model of how crossover homologous recombination occurs during meiosis.[21]
Whether recombination in the DSBR pathway results in chromosomal crossover is determined by how the double Holliday junction is cut, or "resolved". Chromosomal crossover will occur if one Holliday junction is cut on the crossing strand and the other Holliday junction is cut on the non-crossing strand (in Figure 4, along the horizontal purple arrowheads at one Holliday junction and along the vertical orange arrowheads at the other). Alternatively, if the two Holliday junctions are cut on the crossing strands (along the horizontal purple arrowheads at both Holliday junctions in Figure 4), then chromosomes without crossover will be produced.[42]
SDSA pathway
Homologous recombination via the SDSA pathway occurs in cells that divide through mitosis and meiosis and results in non-crossover products. In this model, the invading 3' strand is extended along the recipient DNA duplex by a DNA polymerase, and is released as the Holliday junction between the donor and recipient DNA molecules slides in a process called branch migration. The newly synthesized 3' end of the invading strand is then able to anneal to the other 3' overhang in the damaged chromosome through complementary base pairing. After the strands anneal, a small flap of DNA can sometimes remain. Any such flaps are removed, and the SDSA pathway finishes with the resealing, also known as ligation, of any remaining single-stranded gaps.[43]
During mitosis, the major homologous recombination pathway for repairing DNA double-strand breaks appears to be the SDSA pathway (rather than the DSBR pathway).[44] The SDSA pathway produces non-crossover recombinants (Figure 4). During meiosis non-crossover recombinants also occur frequently and these appear to arise mainly by the SDSA pathway as well.[44][45] Non-crossover recombination events occurring during meiosis likely reflect instances of repair of DNA double-strand damages or other types of DNA damages.
SSA pathway

Figure 5. Recombination via the SSA pathway occurs between two repeat elements (purple) on the same DNA duplex, and results in deletions of genetic material. (Click to view animated diagram in Firefox, Chrome, Safari, or Opera web browsers.)
The single-strand annealing (SSA) pathway of homologous recombination repairs double-strand breaks between two repeat sequences. The SSA pathway is unique in that it does not require a separate similar or identical molecule of DNA, like the DSBR or SDSA pathways of homologous recombination. Instead, the SSA pathway only requires a single DNA duplex, and uses the repeat sequences as the identical sequences that homologous recombination needs for repair. The pathway is relatively simple in concept: after two strands of the same DNA duplex are cut back around the site of the double-strand break, the two resulting 3' overhangs then align and anneal to each other, restoring the DNA as a continuous duplex.[43][46]
As DNA around the double-strand break is cut back, the single-stranded 3' overhangs being produced are coated with the RPA protein, which prevents the 3' overhangs from sticking to themselves.[47] A protein called Rad52 then binds each of the repeat sequences on either side of the break, and aligns them to enable the two complementary repeat sequences to anneal.[47] After annealing is complete, leftover non-homologous flaps of the 3' overhangs are cut away by a set of nucleases, known as Rad1/Rad10, which are brought to the flaps by the Saw1 and Slx4 proteins.[47][48] New DNA synthesis fills in any gaps, and ligation restores the DNA duplex as two continuous strands.[49] The DNA sequence between the repeats is always lost, as is one of the two repeats. The SSA pathway is considered mutagenic since it results in such deletions of genetic material.[43]
BIR pathway
During DNA replication, double-strand breaks can sometimes be encountered at replication forks as DNA helicase unzips the template strand. These defects are repaired in the break-induced replication (BIR) pathway of homologous recombination. The precise molecular mechanisms of the BIR pathway remain unclear. Three proposed mechanisms have strand invasion as an initial step, but they differ in how they model the migration of the D-loop and later phases of recombination.[50]
The BIR pathway can also help to maintain the length of telomeres (regions of DNA at the end of eukaryotic chromosomes) in the absence of (or in cooperation with) telomerase. Without working copies of the enzyme telomerase, telomeres typically shorten with each cycle of mitosis, which eventually blocks cell division and leads to senescence. In budding yeast cells where telomerase has been inactivated through mutations, two types of "survivor" cells have been observed to avoid senescence longer than expected by elongating their telomeres through BIR pathways.[50]
Maintaining telomere length is critical for cell immortalization, a key feature of cancer. Most cancers maintain telomeres by upregulating telomerase. However, in several types of human cancer, a BIR-like pathway helps to sustain some tumors by acting as an alternative mechanism of telomere maintenance.[51] This fact has led scientists to investigate whether such recombination-based mechanisms of telomere maintenance could thwart anti-cancer drugs like telomerase inhibitors.[52]
In bacteria

Figure 6. Crystal structure of a RecA protein filament bound to DNA.[53] A 3' overhang is visible to the right of center.
Homologous recombination is a major DNA repair process in bacteria. It is also important for producing genetic diversity in bacterial populations, although the process differs substantially from meiotic recombination, which repairs DNA damages and brings about diversity in eukaryotic genomes. Homologous recombination has been most studied and is best understood for Escherichia coli.[54] Double-strand DNA breaks in bacteria are repaired by the RecBCD pathway of homologous recombination. Breaks that occur on only one of the two DNA strands, known as single-strand gaps, are thought to be repaired by the RecF pathway.[55] Both the RecBCD and RecF pathways include a series of reactions known as branch migration, in which single DNA strands are exchanged between two intercrossed molecules of duplex DNA, and resolution, in which those two intercrossed molecules of DNA are cut apart and restored to their normal double-stranded state.
RecBCD pathway
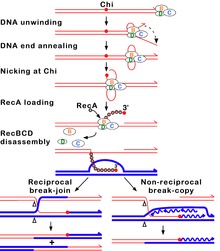
Figure 7A. Molecular model for the RecBCD pathway of recombination. This model is based on reactions of DNA and RecBCD with ATP in excess over Mg2+ ions. Step 1: RecBCD binds to a double-stranded DNA end. Step 2: RecBCD unwinds DNA. RecD is a fast helicase on the 5’-ended strand, and RecB is a slower helicase on the 3'-ended strand (that with an arrowhead) [ref 46 in current Wiki version]. This produces two single-stranded (ss) DNA tails and one ss loop. The loop and tails enlarge as RecBCD moves along the DNA. Step 3: The two tails anneal to produce a second ss DNA loop, and both loops move and grow. Step 4: Upon reaching the Chi hotspot sequence (5' GCTGGTGG 3'; red dot) RecBCD nicks the 3’-ended strand. Further unwinding produces a long 3'-ended ss tail with Chi near its end. Step 5: RecBCD loads RecA protein onto the Chi tail. At some undetermined point, the RecBCD subunits disassemble. Step 6: The RecA-ssDNA complex invades an intact homologous duplex DNA to produce a D-loop, which can be resolved into intact, recombinant DNA in two ways. Step 7: The D-loop is cut and anneals with the gap in the first DNA to produce a Holliday junction. Resolution of the Holliday junction (cutting, swapping of strands, and ligation) at the open arrowheads by some combination of RuvABC and RecG produces two recombinants of reciprocal type. Step 8: The 3' end of the Chi tail primes DNA synthesis, from which a replication fork can be generated. Resolution of the fork at the open arrowheads produces one recombinant (non-reciprocal) DNA, one parental-type DNA, and one DNA fragment.[56]

Figure 7B. Beginning of the RecBCD pathway. This model is based on reactions of DNA and RecBCD with Mg2+ ions in excess over ATP. Step 1: RecBCD binds to a DNA double strand break. Step 2: RecBCD initiates unwinding of the DNA duplex through ATP-dependent helicase activity. Step 3: RecBCD continues its unwinding and moves down the DNA duplex, cleaving the 3' strand much more frequently than the 5' strand. Step 4: RecBCD encounters a Chi sequence and stops digesting the 3' strand; cleavage of the 5' strand is significantly increased. Step 5: RecBCD loads RecA onto the 3' strand. Step 6: RecBCD unbinds from the DNA duplex, leaving a RecA nucleoprotein filament on the 3' tail.[57]
The RecBCD pathway is the main recombination pathway used in many bacteria to repair double-strand breaks in DNA, and the proteins are found in a broad array of bacteria.[58][59][60] These double-strand breaks can be caused by UV light and other radiation, as well as chemical mutagens. Double-strand breaks may also arise by DNA replication through a single-strand nick or gap. Such a situation causes what is known as a collapsed replication fork and is fixed by several pathways of homologous recombination including the RecBCD pathway.[61]
In this pathway, a three-subunit enzyme complex called RecBCD initiates recombination by binding to a blunt or nearly blunt end of a break in double-strand DNA. After RecBCD binds the DNA end, the RecB and RecD subunits begin unzipping the DNA duplex through helicase activity. The RecB subunit also has a nuclease domain, which cuts the single strand of DNA that emerges from the unzipping process. This unzipping continues until RecBCD encounters a specific nucleotide sequence (5'-GCTGGTGG-3') known as a Chi site.[60]
Upon encountering a Chi site, the activity of the RecBCD enzyme changes drastically.[59][56][62] DNA unwinding pauses for a few seconds and then resumes at roughly half the initial speed. This is likely because the slower RecB helicase unwinds the DNA after Chi, rather than the faster RecD helicase, which unwinds the DNA before Chi.[63][64] Recognition of the Chi site also changes the RecBCD enzyme so that it cuts the DNA strand with Chi and begins loading multiple RecA proteins onto the single-stranded DNA with the newly generated 3' end. The resulting RecA-coated nucleoprotein filament then searches out similar sequences of DNA on a homologous chromosome. The search process induces stretching of the DNA duplex, which enhances homology recognition (a mechanism termed conformational proofreading [65][66][67]). Upon finding such a sequence, the single-stranded nucleoprotein filament moves into the homologous recipient DNA duplex in a process called strand invasion.[68] The invading 3' overhang causes one of the strands of the recipient DNA duplex to be displaced, to form a D-loop. If the D-loop is cut, another swapping of strands forms a cross-shaped structure called a Holliday junction.[60] Resolution of the Holliday junction by some combination of RuvABC or RecG can produce two recombinant DNA molecules with reciprocal genetic types, if the two interacting DNA molecules differ genetically. Alternatively, the invading 3’ end near Chi can prime DNA synthesis and form a replication fork. This type of resolution produces only one type of recombinant (non-reciprocal).
RecF pathway
Bacteria appear to use the RecF pathway of homologous recombination to repair single-strand gaps in DNA. When the RecBCD pathway is inactivated by mutations and additional mutations inactivate the SbcCD and ExoI nucleases, the RecF pathway can also repair DNA double-strand breaks.[69] In the RecF pathway the RecQ helicase unwinds the DNA and the RecJ nuclease degrades the strand with a 5' end, leaving the strand with the 3' end intact. RecA protein binds to this strand and is either aided by the RecF, RecO, and RecR proteins or stabilized by them. The RecA nucleoprotein filament then searches for a homologous DNA and exchanges places with the identical or nearly identical strand in the homologous DNA.
Although the proteins and specific mechanisms involved in their initial phases differ, the two pathways are similar in that they both require single-stranded DNA with a 3' end and the RecA protein for strand invasion. The pathways are also similar in their phases of branch migration, in which the Holliday junction slides in one direction, and resolution, in which the Holliday junctions are cleaved apart by enzymes.[70][71] The alternative, non-reciprocal type of resolution may also occur by either pathway.
Branch migration
Immediately after strand invasion, the Holliday junction moves along the linked DNA during the branch migration process. It is in this movement of the Holliday junction that base pairs between the two homologous DNA duplexes are exchanged. To catalyze branch migration, the RuvA protein first recognizes and binds to the Holliday junction and recruits the RuvB protein to form the RuvAB complex. Two sets of the RuvB protein, which each form a ring-shaped ATPase, are loaded onto opposite sides of the Holliday junction, where they act as twin pumps that provide the force for branch migration. Between those two rings of RuvB, two sets of the RuvA protein assemble in the center of the Holliday junction such that the DNA at the junction is sandwiched between each set of RuvA. The strands of both DNA duplexes—the "donor" and the "recipient" duplexes—are unwound on the surface of RuvA as they are guided by the protein from one duplex to the other.[72][73]
Resolution
In the resolution phase of recombination, any Holliday junctions formed by the strand invasion process are cut, thereby restoring two separate DNA molecules. This cleavage is done by RuvAB complex interacting with RuvC, which together form the RuvABC complex. RuvC is an endonuclease that cuts the degenerate sequence 5'-(A/T)TT(G/C)-3'. The sequence is found frequently in DNA, about once every 64 nucleotides.[73] Before cutting, RuvC likely gains access to the Holliday junction by displacing one of the two RuvA tetramers covering the DNA there.[72] Recombination results in either "splice" or "patch" products, depending on how RuvC cleaves the Holliday junction.[73] Splice products are crossover products, in which there is a rearrangement of genetic material around the site of recombination. Patch products, on the other hand, are non-crossover products in which there is no such rearrangement and there is only a "patch" of hybrid DNA in the recombination product.[74]
Facilitating genetic transfer
Homologous recombination is an important method of integrating donor DNA into a recipient organism's genome in horizontal gene transfer, the process by which an organism incorporates foreign DNA from another organism without being the offspring of that organism. Homologous recombination requires incoming DNA to be highly similar to the recipient genome, and so horizontal gene transfer is usually limited to similar bacteria.[75] Studies in several species of bacteria have established that there is a log-linear decrease in recombination frequency with increasing difference in sequence between host and recipient DNA.[76][77][78]
In bacterial conjugation, where DNA is transferred between bacteria through direct cell-to-cell contact, homologous recombination helps integrate foreign DNA into the host genome via the RecBCD pathway. The RecBCD enzyme promotes recombination after DNA is converted from single-strand DNA–in which form it originally enters the bacterium–to double-strand DNA during replication. The RecBCD pathway is also essential for the final phase of transduction, a type of horizontal gene transfer in which DNA is transferred from one bacterium to another by a virus. Foreign, bacterial DNA is sometimes misincorporated in the capsid head of bacteriophage virus particles as DNA is packaged into new bacteriophages during viral replication. When these new bacteriophages infect other bacteria, DNA from the previous host bacterium is injected into the new bacterial host as double-strand DNA. The RecBCD enzyme then incorporates this double-strand DNA into the genome of the new bacterial host.[60]
Bacterial transformation
Natural bacterial transformation involves the transfer of DNA from a donor bacterium to a recipient bacterium, where both donor and recipient are ordinarily of the same species. Transformation, unlike bacterial conjugation and transduction, depends on numerous bacterial gene products that specifically interact to perform this process.[79] Thus transformation is clearly a bacterial adaptation for DNA transfer. In order for a bacterium to bind, take up and integrate donor DNA into its resident chromosome by homologous recombination, it must first enter a special physiological state termed competence. The RecA/Rad51/DMC1 gene family plays a central role in homologous recombination during bacterial transformation as it does during eukaryotic meiosis and mitosis. For instance, the RecA protein is essential for transformation in Bacillus subtilis and Streptococcus pneumoniae,[80] and expression of the RecA gene is induced during the development of competence for transformation in these organisms.
As part of the transformation process, the RecA protein interacts with entering single-stranded DNA (ssDNA) to form RecA/ssDNA nucleofilaments that scan the resident chromosome for regions of homology and bring the entering ssDNA to the corresponding region, where strand exchange and homologous recombination occur.[81] Thus the process of homologous recombination during bacterial transformation has fundamental similarities to homologous recombination during meiosis.
In viruses
Homologous recombination occurs in several groups of viruses. In DNA viruses such as herpesvirus, recombination occurs through a break-and-rejoin mechanism like in bacteria and eukaryotes.[82] There is also evidence for recombination in some RNA viruses, specifically positive-sense ssRNA viruses like retroviruses, picornaviruses, and coronaviruses. There is controversy over whether homologous recombination occurs in negative-sense ssRNA viruses like influenza.[83]
In RNA viruses, homologous recombination can be either precise or imprecise. In the precise type of RNA-RNA recombination, there is no difference between the two parental RNA sequences and the resulting crossover RNA region. Because of this, it is often difficult to determine the location of crossover events between two recombining RNA sequences. In imprecise RNA homologous recombination, the crossover region has some difference with the parental RNA sequences – caused by either addition, deletion, or other modification of nucleotides. The level of precision in crossover is controlled by the sequence context of the two recombining strands of RNA: sequences rich in adenine and uracil decrease crossover precision.[84][85]
Homologous recombination is important in facilitating viral evolution.[84][86] For example, if the genomes of two viruses with different disadvantageous mutations undergo recombination, then they may be able to regenerate a fully functional genome. Alternatively, if two similar viruses have infected the same host cell, homologous recombination can allow those two viruses to swap genes and thereby evolve more potent variations of themselves.[86]
Homologous recombination is the proposed mechanism whereby the DNA virus human herpesvirus-6 integrates into human telomeres.[87]
When two or more viruses, each containing lethal genomic damage, infect the same host cell, the virus genomes can often pair with each other and undergo homologous recombinational repair to produce viable progeny. This process, known as multiplicity reactivation, has been studied in several bacteriophages, including phage T4.[88] Enzymes employed in recombinational repair in phage T4 are functionally homologous to enzymes employed in bacterial and eukaryotic recombinational repair.[89] In particular, with regard to a gene necessary for the strand exchange reaction, a key step in homologous recombinational repair, there is functional homology from viruses to humans (i. e. uvsX in phage T4; recA in E. coli and other bacteria, and rad51 and dmc1 in yeast and other eukaryotes, including humans).[90] Multiplicity reactivation has also been demonstrated in numerous pathogenic viruses.[91]
Effects of dysfunction
Without proper homologous recombination, chromosomes often incorrectly align for the first phase of cell division in meiosis. This causes chromosomes to fail to properly segregate in a process called nondisjunction. In turn, nondisjunction can cause sperm and ova to have too few or too many chromosomes. Down's syndrome, which is caused by an extra copy of chromosome 21, is one of many abnormalities that result from such a failure of homologous recombination in meiosis.[73][92]
Deficiencies in homologous recombination have been strongly linked to cancer formation in humans. For example, each of the cancer-related diseases Bloom's syndrome, Werner's syndrome and Rothmund-Thomson syndrome are caused by malfunctioning copies of RecQ helicase genes involved in the regulation of homologous recombination: BLM, WRN and RECQL4, respectively.[93] In the cells of Bloom's syndrome patients, who lack a working copy of the BLM protein, there is an elevated rate of homologous recombination.[94] Experiments in mice deficient in BLM have suggested that the mutation gives rise to cancer through a loss of heterozygosity caused by increased homologous recombination.[95] A loss in heterozygosity refers to the loss of one of two versions—or alleles—of a gene. If one of the lost alleles helps to suppress tumors, like the gene for the retinoblastoma protein for example, then the loss of heterozygosity can lead to cancer.[96]:1236
Decreased rates of homologous recombination cause inefficient DNA repair,[96]:310 which can also lead to cancer.[97] This is the case with BRCA1 and BRCA2, two similar tumor suppressor genes whose malfunctioning has been linked with considerably increased risk for breast and ovarian cancer. Cells missing BRCA1 and BRCA2 have a decreased rate of homologous recombination and increased sensitivity to ionizing radiation, suggesting that decreased homologous recombination leads to increased susceptibility to cancer.[97] Because the only known function of BRCA2 is to help initiate homologous recombination, researchers have speculated that more detailed knowledge of BRCA2's role in homologous recombination may be the key to understanding the causes of breast and ovarian cancer.[97]
Tumours with a homologous recombination deficiency (including BRCA defects) are described as HRD-positive.[98]
Evolutionary conservation

Figure 8. Protein domains in homologous recombination-related proteins are conserved across the three main groups of life: archaea, bacteria and eukaryotes.
While the pathways can mechanistically vary, the ability of organisms to perform homologous recombination is universally conserved across all domains of life.[99] Based on the similarity of their amino acid sequences, homologs of a number of proteins can be found in multiple domains of life indicating that they evolved a long time ago, and have since diverged from common ancestral proteins.[99]
RecA recombinase family members are found in almost all organisms with RecA in bacteria, Rad51 and DMC1 in eukaryotes, RadA in archaea, and UvsX in T4 phage.[100]
Related single stranded binding proteins that are important for homologous recombination, and many other processes, are also found in all domains of life.[101]
Rad54, Mre11, Rad50, and a number of other proteins are also found in both archaea and eukaryotes.[99][100][102]
The RecA recombinase family
The proteins of the RecA recombinase family of proteins are thought to be descended from a common ancestral recombinase.[99] The RecA recombinase family contains RecA protein from bacteria, the Rad51 and Dmc1 proteins from eukaryotes, and RadA from archaea, and the recombinase paralog proteins. Studies modeling the evolutionary relationships between the Rad51, Dmc1 and RadA proteins indicate that they are monophyletic, or that they share a common molecular ancestor.[99] Within this protein family, Rad51 and Dmc1 are grouped together in a separate clade from RadA. One of the reasons for grouping these three proteins together is that they all possess a modified helix-turn-helix motif, which helps the proteins bind to DNA, toward their N-terminal ends.[99] An ancient gene duplication event of a eukaryotic RecA gene and subsequent mutation has been proposed as a likely origin of the modern RAD51 and DMC1 genes.[99]
The proteins generally share a long conserved region known as the RecA/Rad51 domain. Within this protein domain are two sequence motifs, Walker A motif and Walker B motif. The Walker A and B motifs allow members of the RecA/Rad51 protein family to engage in ATP binding and ATP hydrolysis.[99][103]
Meiosis-specific proteins
The discovery of Dmc1 in several species of Giardia, one of the earliest protists to diverge as a eukaryote, suggests that meiotic homologous recombination—and thus meiosis itself—emerged very early in eukaryotic evolution.[104] In addition to research on Dmc1, studies on the Spo11 protein have provided information on the origins of meiotic recombination.[105] Spo11, a type II topoisomerase, can initiate homologous recombination in meiosis by making targeted double-strand breaks in DNA.[24]Phylogenetic trees based on the sequence of genes similar to SPO11 in animals, fungi, plants, protists and archaea have led scientists to believe that the version Spo11 currently in eukaryotes emerged in the last common ancestor of eukaryotes and archaea.[105]
Technological applications
Gene targeting

Figure 9. As a developing embryo, this chimeric mouse had the agouti coat color gene introduced into its DNA via gene targeting. Its offspring are homozygous for the agouti gene.
Many methods for introducing DNA sequences into organisms to create recombinant DNA and genetically modified organisms use the process of homologous recombination.[106] Also called gene targeting, the method is especially common in yeast and mouse genetics. The gene targeting method in knockout mice uses mouse embryonic stem cells to deliver artificial genetic material (mostly of therapeutic interest), which represses the target gene of the mouse by the principle of homologous recombination. The mouse thereby acts as a working model to understand the effects of a specific mammalian gene. In recognition of their discovery of how homologous recombination can be used to introduce genetic modifications in mice through embryonic stem cells, Mario Capecchi, Martin Evans and Oliver Smithies were awarded the 2007 Nobel Prize for Physiology or Medicine.[107]
Advances in gene targeting technologies which hijack the homologous recombination mechanics of cells are now leading to the development of a new wave of more accurate, isogenic human disease models. These engineered human cell models are thought to more accurately reflect the genetics of human diseases than their mouse model predecessors. This is largely because mutations of interest are introduced into endogenous genes, just as they occur in the real patients, and because they are based on human genomes rather than rat genomes. Furthermore, certain technologies enable the knock-in of a particular mutation rather than just knock-outs associated with older gene targeting technologies.
Protein engineering
Protein engineering with homologous recombination develops chimeric proteins by swapping fragments between two parental proteins. These techniques exploit the fact that recombination can introduce a high degree of sequence diversity while preserving a protein's ability to fold into its tertiary structure, or three-dimensional shape.[108] This stands in contrast to other protein engineering techniques, like random point mutagenesis, in which the probability of maintaining protein function declines exponentially with increasing amino acid substitutions.[109] The chimeras produced by recombination techniques are able to maintain their ability to fold because their swapped parental fragments are structurally and evolutionarily conserved. These recombinable "building blocks" preserve structurally important interactions like points of physical contact between different amino acids in the protein's structure. Computational methods like SCHEMA and statistical coupling analysis can be used to identify structural subunits suitable for recombination.[110][111][112]
Techniques that rely on homologous recombination have been used to engineer new proteins.[110] In a study published in 2007, researchers were able to create chimeras of two enzymes involved in the biosynthesis of isoprenoids, a diverse class of compounds including hormones, visual pigments and certain pheromones. The chimeric proteins acquired an ability to catalyze an essential reaction in isoprenoid biosynthesis—one of the most diverse pathways of biosynthesis found in nature—that was absent in the parent proteins.[113] Protein engineering through recombination has also produced chimeric enzymes with new function in members of a group of proteins known as the cytochrome P450 family,[114] which in humans is involved in detoxifying foreign compounds like drugs, food additives and preservatives.[21]
Cancer therapy
Cancer cells with BRCA mutations have deficiencies in homologous recombination, and drugs to exploit those deficiencies have been developed and used successfully in clinical trials.[115][116]Olaparib, a PARP1 inhibitor, shrunk or stopped the growth of tumors from breast, ovarian and prostate cancers caused by mutations in the BRCA1 or BRCA2 genes, which are necessary for HR. When BRCA1 or BRCA2 is absent, other types of DNA repair mechanisms must compensate for the deficiency of HR, such as base-excision repair (BER) for stalled replication forks or non-homologous end joining (NHEJ) for double strand breaks.[115] By inhibiting BER in an HR-deficient cell, olaparib applies the concept of synthetic lethality to specifically target cancer cells. While PARP1 inhibitors represent a novel approach to cancer therapy, researchers have cautioned that they may prove insufficient for treating late-stage metastatic cancers.[115] Cancer cells can become resistant to a PARP1 inhibitor if they undergo deletions of mutations in BRCA2, undermining the drug's synthetic lethality by restoring cancer cells' ability to repair DNA by HR.[117]
See also
- Chromosomal crossover
- Homology directed repair
References
^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P, et al. (2002). "Chapter 5: DNA Replication, Repair, and Recombination". Molecular Biology of the Cell (4th ed.). New York: Garland Science. p. 845. ISBN 978-0-8153-3218-3. OCLC 145080076..mw-parser-output cite.citationfont-style:inherit.mw-parser-output .citation qquotes:"""""""'""'".mw-parser-output .citation .cs1-lock-free abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .citation .cs1-lock-limited a,.mw-parser-output .citation .cs1-lock-registration abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .citation .cs1-lock-subscription abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registrationcolor:#555.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration spanborder-bottom:1px dotted;cursor:help.mw-parser-output .cs1-ws-icon abackground:url("//upload.wikimedia.org/wikipedia/commons/thumb/4/4c/Wikisource-logo.svg/12px-Wikisource-logo.svg.png")no-repeat;background-position:right .1em center.mw-parser-output code.cs1-codecolor:inherit;background:inherit;border:inherit;padding:inherit.mw-parser-output .cs1-hidden-errordisplay:none;font-size:100%.mw-parser-output .cs1-visible-errorfont-size:100%.mw-parser-output .cs1-maintdisplay:none;color:#33aa33;margin-left:0.3em.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-formatfont-size:95%.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-leftpadding-left:0.2em.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-rightpadding-right:0.2em
^ Capecchi MR (June 1989). "Altering the genome by homologous recombination". Science. 244 (4910): 1288–92. doi:10.1126/science.2660260. PMID 2660260.
^ Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS (1985-09-19). "Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination". Nature. 317 (6034): 230–4. doi:10.1038/317230a0. PMID 2995814.
^ Orr-Weaver TL, Szostak JW, Rothstein RJ (October 1981). "Yeast transformation: a model system for the study of recombination". Proceedings of the National Academy of Sciences of the United States of America. 78 (10): 6354–8. doi:10.1073/pnas.78.10.6354. PMC 349037. PMID 6273866.
^ Orr-Weaver TL, Szostak JW (July 1983). "Yeast recombination: the association between double-strand gap repair and crossing-over". Proceedings of the National Academy of Sciences of the United States of America. 80 (14): 4417–21. doi:10.1073/pnas.80.14.4417. PMC 384049. PMID 6308623.
^ ab Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW (May 1983). "The double-strand-break repair model for recombination". Cell. 33 (1): 25–35. doi:10.1016/0092-8674(83)90331-8. PMID 6380756.
^ Resnick MA (June 1976). "The repair of double-strand breaks in DNA; a model involving recombination". Journal of Theoretical Biology. 59 (1): 97–106. doi:10.1016/s0022-5193(76)80025-2. PMID 940351.
^ Jasin M, Rothstein R (November 2013). "Repair of strand breaks by homologous recombination". Cold Spring Harbor Perspectives in Biology. 5 (11): a012740. doi:10.1101/cshperspect.a012740. PMC 3809576. PMID 24097900.
^ Bateson P (August 2002). "William Bateson: a biologist ahead of his time" (PDF). Journal of Genetics. 81 (2): 49–58. doi:10.1007/BF02715900. PMID 12532036.
^ "Reginald Crundall Punnett". NAHSTE, University of Edinburgh. Retrieved 3 July 2010.
^ Lobo I, Shaw K (2008). "Thomas Hunt Morgan, genetic recombination, and gene mapping". Nature Education. 1 (1).
^ ab Coe E, Kass LB (May 2005). "Proof of physical exchange of genes on the chromosomes". Proceedings of the National Academy of Sciences of the United States of America. 102 (19): 6641–6. doi:10.1073/pnas.0407340102. PMC 1100733. PMID 15867161.
^ Creighton HB, McClintock B (August 1931). "A Correlation of Cytological and Genetical Crossing-Over in Zea Mays". Proceedings of the National Academy of Sciences of the United States of America. 17 (8): 492–7. doi:10.1073/pnas.17.8.492. PMC 1076098. PMID 16587654.
^ Stern, C (1931). "Zytologisch-genetische untersuchungen alsbeweise fur die Morgansche theorie des faktoraustauschs". Biol. Zentbl. 51: 547–587.
^ "The development of bacterial genetics". US National Library of Medicine. Retrieved 3 July 2010.
^ "The Nobel Prize in Physiology or Medicine 1958". Nobelprize.org. Retrieved 3 July 2010.
^ abc Haber JE, Ira G, Malkova A, Sugawara N (January 2004). "Repairing a double-strand chromosome break by homologous recombination: revisiting Robin Holliday's model". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 359 (1441): 79–86. doi:10.1098/rstb.2003.1367. PMC 1693306. PMID 15065659.
^ Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J (2000). "12.5: Recombination between Homologous DNA Sites: Double-Strand Breaks in DNA Initiate Recombination". Molecular Cell Biology (4th ed.). W. H. Freeman and Company. ISBN 978-0-7167-3136-8.
^ Griffiths A, et al. (1999). "8: Chromosome Mutations: Chromosomal Rearrangements". Modern Genetic Analysis. W. H. Freeman and Company. ISBN 978-0-7167-3118-4.
^ Khanna KK, Jackson SP (March 2001). "DNA double-strand breaks: signaling, repair and the cancer connection". Nature Genetics. 27 (3): 247–54. doi:10.1038/85798. PMID 11242102.
^ abc Nelson DL, Cox MM (2005). Principles of Biochemistry (4th ed.). Freeman. pp. 980–981. ISBN 978-0-7167-4339-2.
^ Marcon E, Moens PB (August 2005). "The evolution of meiosis: recruitment and modification of somatic DNA-repair proteins". BioEssays. 27 (8): 795–808. doi:10.1002/bies.20264. PMID 16015600.
^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008). Molecular Biology of the Cell (5th ed.). Garland Science. p. 305. ISBN 978-0-8153-4105-5.
^ ab Keeney S, Giroux CN, Kleckner N (February 1997). "Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family". Cell. 88 (3): 375–84. doi:10.1016/S0092-8674(00)81876-0. PMID 9039264.
^ Longhese MP, Bonetti D, Guerini I, Manfrini N, Clerici M (September 2009). "DNA double-strand breaks in meiosis: checking their formation, processing and repair". DNA Repair. 8 (9): 1127–38. doi:10.1016/j.dnarep.2009.04.005. PMID 19464965.
^ Cahill LP, Mariana JC, Mauléon P (January 1979). "Total follicular populations in ewes of high and low ovulation rates". Journal of Reproduction and Fertility. 55 (1): 27–36. doi:10.1371/journal.pbio.0020192. PMC 423159. PMID 423159.
^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008). Molecular Biology of the Cell (5th ed.). Garland Science. p. 303. ISBN 978-0-8153-4105-5.
^ ab Shrivastav M, De Haro LP, Nickoloff JA (January 2008). "Regulation of DNA double-strand break repair pathway choice". Cell Research. 18 (1): 134–47. doi:10.1038/cr.2007.111. PMID 18157161.
^ ab Mimitou EP, Symington LS (May 2009). "Nucleases and helicases take center stage in homologous recombination". Trends in Biochemical Sciences. 34 (5): 264–72. doi:10.1016/j.tibs.2009.01.010. PMID 19375328.
^ Huertas P, Cortés-Ledesma F, Sartori AA, Aguilera A, Jackson SP (October 2008). "CDK targets Sae2 to control DNA-end resection and homologous recombination". Nature. 455 (7213): 689–92. doi:10.1038/nature07215. PMC 2635538. PMID 18716619.
^ Liu B, Yip RK, Zhou Z (2012). "Chromatin remodeling, DNA damage repair and aging". Curr. Genomics. 13 (7): 533–47. doi:10.2174/138920212803251373. PMC 3468886. PMID 23633913.
^ abcd Sellou H, Lebeaupin T, Chapuis C, Smith R, Hegele A, Singh HR, Kozlowski M, Bultmann S, Ladurner AG, Timinszky G, Huet S (2016). "The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage". Mol. Biol. Cell. 27 (24): 3791–3799. doi:10.1091/mbc.E16-05-0269. PMC 5170603. PMID 27733626.
^ ab Van Meter M, Simon M, Tombline G, May A, Morello TD, Hubbard BP, Bredbenner K, Park R, Sinclair DA, Bohr VA, Gorbunova V, Seluanov A (2016). "JNK Phosphorylates SIRT6 to Stimulate DNA Double-Strand Break Repair in Response to Oxidative Stress by Recruiting PARP1 to DNA Breaks". Cell Rep. 16 (10): 2641–50. doi:10.1016/j.celrep.2016.08.006. PMC 5089070. PMID 27568560.
^ ab Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG (2008). "PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites". J. Biol. Chem. 283 (2): 1197–208. doi:10.1074/jbc.M706734200. PMID 18025084.
^ abc Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM (1998). "DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139". J. Biol. Chem. 273 (10): 5858–68. doi:10.1074/jbc.273.10.5858. PMID 9488723.
^ Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J (2007). "RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins". Cell. 131 (5): 887–900. doi:10.1016/j.cell.2007.09.040. PMID 18001824.
^ Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH, Khanna KK, van Attikum H, Mailand N, Dantuma NP (2012). "A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure". EMBO J. 31 (11): 2511–27. doi:10.1038/emboj.2012.104. PMC 3365417. PMID 22531782.
^ abcd Sung P, Klein H (October 2006). "Mechanism of homologous recombination: mediators and helicases take on regulatory functions". Nature Reviews Molecular Cell Biology. 7 (10): 739–50. doi:10.1038/nrm2008. PMID 16926856.
^ Wold MS (1997). "Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism". Annual Review of Biochemistry. 66: 61–92. doi:10.1146/annurev.biochem.66.1.61. PMID 9242902.
^ McMahill MS, Sham CW, Bishop DK (November 2007). "Synthesis-dependent strand annealing in meiosis". PLoS Biology. 5 (11): e299. doi:10.1371/journal.pbio.0050299. PMC 2062477. PMID 17988174.
^ Bärtsch S, Kang LE, Symington LS (February 2000). "RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates". Molecular and Cellular Biology. 20 (4): 1194–205. doi:10.1128/MCB.20.4.1194-1205.2000. PMC 85244. PMID 10648605.
^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008). Molecular Biology of the Cell (5th ed.). Garland Science. pp. 312–313. ISBN 978-0-8153-4105-5.
^ abc Helleday T, Lo J, van Gent DC, Engelward BP (July 2007). "DNA double-strand break repair: from mechanistic understanding to cancer treatment". DNA Repair. 6 (7): 923–35. doi:10.1016/j.dnarep.2007.02.006. PMID 17363343.
^ ab Andersen SL, Sekelsky J (December 2010). "Meiotic versus mitotic recombination: two different routes for double-strand break repair: the different functions of meiotic versus mitotic DSB repair are reflected in different pathway usage and different outcomes". BioEssays. 32 (12): 1058–66. doi:10.1002/bies.201000087. PMC 3090628. PMID 20967781.
^ Allers T, Lichten M (July 2001). "Differential timing and control of noncrossover and crossover recombination during meiosis". Cell. 106 (1): 47–57. doi:10.1016/s0092-8674(01)00416-0. PMID 11461701.
^ Haber lab. "Single-strand annealing". "Brandeis University". Retrieved 3 July 2010.
^ abc Lyndaker AM, Alani E (March 2009). "A tale of tails: insights into the coordination of 3' end processing during homologous recombination". BioEssays. 31 (3): 315–21. doi:10.1002/bies.200800195. PMC 2958051. PMID 19260026.
^ Mimitou EP, Symington LS (September 2009). "DNA end resection: many nucleases make light work". DNA Repair. 8 (9): 983–95. doi:10.1016/j.dnarep.2009.04.017. PMC 2760233. PMID 19473888.
^ Pâques F, Haber JE (June 1999). "Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae". Microbiology and Molecular Biology Reviews. 63 (2): 349–404. PMC 98970. PMID 10357855.
^ ab McEachern MJ, Haber JE (2006). "Break-induced replication and recombinational telomere elongation in yeast". Annual Review of Biochemistry. 75: 111–35. doi:10.1146/annurev.biochem.74.082803.133234. PMID 16756487.
^ Morrish TA, Greider CW (January 2009). Haber JE, ed. "Short telomeres initiate telomere recombination in primary and tumor cells". PLoS Genetics. 5 (1): e1000357. doi:10.1371/journal.pgen.1000357. PMC 2627939. PMID 19180191.
^ Muntoni A, Reddel RR (October 2005). "The first molecular details of ALT in human tumor cells". Human Molecular Genetics. 14 Spec No. 2 (Review Issue 2): R191–6. doi:10.1093/hmg/ddi266. PMID 16244317.
^ PDB: 3cmt; Chen Z, Yang H, Pavletich NP (May 2008). "Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures". Nature. 453 (7194): 489–4. doi:10.1038/nature06971. PMID 18497818.
^ Kowalczykowski SC, Dixon DA, Eggleston AK, Lauder SD, Rehrauer WM (September 1994). "Biochemistry of homologous recombination in Escherichia coli". Microbiological Reviews. 58 (3): 401–65. PMC 372975. PMID 7968921.
^ Rocha EP, Cornet E, Michel B (August 2005). "Comparative and evolutionary analysis of the bacterial homologous recombination systems". PLoS Genetics. 1 (2): e15. doi:10.1371/journal.pgen.0010015. PMC 1193525. PMID 16132081.
^ ab Amundsen SK, Taylor AF, Reddy M, Smith GR (December 2007). "Intersubunit signaling in RecBCD enzyme, a complex protein machine regulated by Chi hot spots". Genes & Development. 21 (24): 3296–307. doi:10.1101/gad.1605807. PMC 2113030. PMID 18079176.
^ Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DB (November 2004). "Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks" (PDF). Nature. 432 (7014): 187–93. doi:10.1038/nature02988. PMID 15538360. Archived from the original (PDF) on 2004-05-25.
^ Cromie GA (August 2009). "Phylogenetic ubiquity and shuffling of the bacterial RecBCD and AddAB recombination complexes". Journal of Bacteriology. 191 (16): 5076–84. doi:10.1128/JB.00254-09. PMC 2725590. PMID 19542287.
^ ab Smith GR (June 2012). "How RecBCD enzyme and Chi promote DNA break repair and recombination: a molecular biologist's view". Microbiology and Molecular Biology Reviews. 76 (2): 217–28. doi:10.1128/MMBR.05026-11. PMC 3372252. PMID 22688812.
^ abcd Dillingham MS, Kowalczykowski SC (December 2008). "RecBCD enzyme and the repair of double-stranded DNA breaks". Microbiology and Molecular Biology Reviews. 72 (4): 642–71, Table of Contents. doi:10.1128/MMBR.00020-08. PMC 2593567. PMID 19052323.
^ Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R (July 2007). "Recombination proteins and rescue of arrested replication forks". DNA Repair. 6 (7): 967–80. doi:10.1016/j.dnarep.2007.02.016. PMID 17395553.
^ Spies M, Bianco PR, Dillingham MS, Handa N, Baskin RJ, Kowalczykowski SC (September 2003). "A molecular throttle: the recombination hotspot chi controls DNA translocation by the RecBCD helicase". Cell. 114 (5): 647–54. doi:10.1016/S0092-8674(03)00681-0. PMID 13678587.
^ Taylor AF, Smith GR (June 2003). "RecBCD enzyme is a DNA helicase with fast and slow motors of opposite polarity". Nature. 423 (6942): 889–93. doi:10.1038/nature01674. PMID 12815437.
^ Spies M, Amitani I, Baskin RJ, Kowalczykowski SC (November 2007). "RecBCD enzyme switches lead motor subunits in response to chi recognition". Cell. 131 (4): 694–705. doi:10.1016/j.cell.2007.09.023. PMC 2151923. PMID 18022364.
^ Savir Y, Tlusty T (November 2010). "RecA-mediated homology search as a nearly optimal signal detection system" (PDF). Molecular Cell. 40 (3): 388–96. doi:10.1016/j.molcel.2010.10.020. PMID 21070965.
^ Rambo RP, Williams GJ, Tainer JA (November 2010). "Achieving fidelity in homologous recombination despite extreme complexity: informed decisions by molecular profiling" (PDF). Molecular Cell. 40 (3): 347–8. doi:10.1016/j.molcel.2010.10.032. PMC 3003302. PMID 21070960.
^ De Vlaminck I, van Loenhout MT, Zweifel L, den Blanken J, Hooning K, Hage S, Kerssemakers J, Dekker C (June 2012). "Mechanism of homology recognition in DNA recombination from dual-molecule experiments". Molecular Cell. 46 (5): 616–24. doi:10.1016/j.molcel.2012.03.029. PMID 22560720.
^ Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2008). Molecular Biology of the Cell (5th ed.). Garland Science. p. 307. ISBN 978-0-8153-4105-5.
^ Morimatsu K, Kowalczykowski SC (May 2003). "RecFOR proteins load RecA protein onto gapped DNA to accelerate DNA strand exchange: a universal step of recombinational repair". Molecular Cell. 11 (5): 1337–47. doi:10.1016/S1097-2765(03)00188-6. PMID 12769856.
^ Hiom K (July 2009). "DNA repair: common approaches to fixing double-strand breaks". Current Biology. 19 (13): R523–5. doi:10.1016/j.cub.2009.06.009. PMID 19602417.
^ Handa N, Morimatsu K, Lovett ST, Kowalczykowski SC (May 2009). "Reconstitution of initial steps of dsDNA break repair by the RecF pathway of E. coli". Genes & Development. 23 (10): 1234–45. doi:10.1101/gad.1780709. PMC 2685532. PMID 19451222.
^ ab West SC (June 2003). "Molecular views of recombination proteins and their control". Nature Reviews Molecular Cell Biology. 4 (6): 435–45. doi:10.1038/nrm1127. PMID 12778123.
^ abcd Watson JD, Baker TA, Bell SP, Gann A, Levine M, Losick R (2003). Molecular Biology of the Gene (5th ed.). Pearson/Benjamin Cummings. pp. 259–291. ISBN 978-0-8053-4635-0.
^ Gumbiner-Russo LM, Rosenberg SM (28 November 2007). Sandler S, ed. "Physical analyses of E. coli heteroduplex recombination products in vivo: on the prevalence of 5' and 3' patches". PLOS ONE. 2 (11): e1242. doi:10.1371/journal.pone.0001242. PMC 2082072. PMID 18043749.
^ Thomas CM, Nielsen KM (September 2005). "Mechanisms of, and barriers to, horizontal gene transfer between bacteria" (PDF). Nature Reviews. Microbiology. 3 (9): 711–21. doi:10.1038/nrmicro1234. PMID 16138099. Archived from the original (PDF) on 2010-06-01.
^ Vulić M, Dionisio F, Taddei F, Radman M (September 1997). "Molecular keys to speciation: DNA polymorphism and the control of genetic exchange in enterobacteria". Proceedings of the National Academy of Sciences of the United States of America. 94 (18): 9763–7. doi:10.1073/pnas.94.18.9763. PMC 23264. PMID 9275198.
^ Majewski J, Cohan FM (January 1998). "The effect of mismatch repair and heteroduplex formation on sexual isolation in Bacillus". Genetics. 148 (1): 13–8. PMC 1459767. PMID 9475717.
^ Majewski J, Zawadzki P, Pickerill P, Cohan FM, Dowson CG (February 2000). "Barriers to genetic exchange between bacterial species: Streptococcus pneumoniae transformation". Journal of Bacteriology. 182 (4): 1016–23. doi:10.1128/JB.182.4.1016-1023.2000. PMC 94378. PMID 10648528.
^ Chen I, Dubnau D (March 2004). "DNA uptake during bacterial transformation". Nature Reviews. Microbiology. 2 (3): 241–9. doi:10.1038/nrmicro844. PMID 15083159.
^ Claverys JP, Martin B, Polard P (May 2009). "The genetic transformation machinery: composition, localization, and mechanism". FEMS Microbiology Reviews. 33 (3): 643–56. doi:10.1111/j.1574-6976.2009.00164.x. PMID 19228200.
^ Kidane D, Graumann PL (July 2005). "Intracellular protein and DNA dynamics in competent Bacillus subtilis cells". Cell. 122 (1): 73–84. doi:10.1016/j.cell.2005.04.036. PMID 16009134.
^ Fleischmann Jr WR (1996). "43". Medical Microbiology (4th ed.). University of Texas Medical Branch at Galveston. ISBN 978-0-9631172-1-2.
^ Boni MF, de Jong MD, van Doorn HR, Holmes EC (3 May 2010). Martin DP, ed. "Guidelines for identifying homologous recombination events in influenza A virus". PLOS ONE. 5 (5): e10434. doi:10.1371/journal.pone.0010434. PMC 2862710. PMID 20454662.
^ ab Nagy PD, Bujarski JJ (January 1996). "Homologous RNA recombination in brome mosaic virus: AU-rich sequences decrease the accuracy of crossovers". Journal of Virology. 70 (1): 415–26. PMC 189831. PMID 8523555.
^ Chetverin AB (October 1999). "The puzzle of RNA recombination". FEBS Letters. 460 (1): 1–5. doi:10.1016/S0014-5793(99)01282-X. PMID 10571050.
^ ab Roossinck MJ (September 1997). "Mechanisms of plant virus evolution". Annual Review of Phytopathology. 35: 191–209. doi:10.1146/annurev.phyto.35.1.191. PMID 15012521.
^ Arbuckle JH, Medveczky PG (August 2011). "The molecular biology of human herpesvirus-6 latency and telomere integration". Microbes and Infection / Institut Pasteur. 13 (8–9): 731–41. doi:10.1016/j.micinf.2011.03.006. PMC 3130849. PMID 21458587.
^ Bernstein C (March 1981). "Deoxyribonucleic acid repair in bacteriophage". Microbiological Reviews. 45 (1): 72–98. PMC 281499. PMID 6261109.
^ Bernstein C, Bernstein H (2001). DNA repair in bacteriophage. In: Nickoloff JA, Hoekstra MF (Eds.) DNA Damage and Repair, Vol.3. Advances from Phage to Humans. Humana Press, Totowa, NJ, pp. 1–19.
ISBN 978-0896038035
^ Story RM, Bishop DK, Kleckner N, Steitz TA (March 1993). "Structural relationship of bacterial RecA proteins to recombination proteins from bacteriophage T4 and yeast". Science. 259 (5103): 1892–6. doi:10.1126/science.8456313. PMID 8456313.
^ Michod RE, Bernstein H, Nedelcu AM (May 2008). "Adaptive value of sex in microbial pathogens". Infection, Genetics and Evolution. 8 (3): 267–85. doi:10.1016/j.meegid.2008.01.002. PMID 18295550.
http://www.hummingbirds.arizona.edu/Faculty/Michod/Downloads/IGE%20review%20sex.pdf
^ Lamb NE, Yu K, Shaffer J, Feingold E, Sherman SL (January 2005). "Association between maternal age and meiotic recombination for trisomy 21". American Journal of Human Genetics. 76 (1): 91–9. doi:10.1086/427266. PMC 1196437. PMID 15551222.
^ Cold Spring Harbor Laboratory (2007). "Human RecQ Helicases, Homologous Recombination And Genomic Instability". ScienceDaily. Retrieved 3 July 2010.
^ Modesti M, Kanaar R (2001). "Homologous recombination: from model organisms to human disease". Genome Biology. 2 (5): REVIEWS1014. doi:10.1186/gb-2001-2-5-reviews1014. PMC 138934. PMID 11387040.
^ Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, Youssoufian H, Vogel H, Schultz RA, Bradley A (December 2000). "Cancer predisposition caused by elevated mitotic recombination in Bloom mice". Nature Genetics. 26 (4): 424–9. doi:10.1038/82548. PMID 11101838.
^ ab Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2007). Molecular Biology of the Cell (5th ed.). Garland Science. ISBN 978-0-8153-4110-9.
^ abc Powell SN, Kachnic LA (September 2003). "Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation". Oncogene. 22 (37): 5784–91. doi:10.1038/sj.onc.1206678. PMID 12947386.
^ Use of homologous recombination deficiency (HRD) score to enrich for niraparib sensitive high grade ovarian tumors.
^ abcdefgh Lin Z, Kong H, Nei M, Ma H (July 2006). "Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer". Proceedings of the National Academy of Sciences of the United States of America. 103 (27): 10328–33. doi:10.1073/pnas.0604232103. PMC 1502457. PMID 16798872.
^ ab Haseltine CA, Kowalczykowski SC (May 2009). "An archaeal Rad54 protein remodels DNA and stimulates DNA strand exchange by RadA". Nucleic Acids Research. 37 (8): 2757–70. doi:10.1093/nar/gkp068. PMC 2677860. PMID 19282450.
^ Rolfsmeier ML, Haseltine CA (March 2010). "The single-stranded DNA binding protein of Sulfolobus solfataricus acts in the presynaptic step of homologous recombination". Journal of Molecular Biology. 397 (1): 31–45. doi:10.1016/j.jmb.2010.01.004. PMID 20080104.
^ Huang Q, Liu L, Liu J, Ni J, She Q, Shen Y (2015). "Efficient 5'-3' DNA end resection by HerA and NurA is essential for cell viability in the crenarchaeon Sulfolobus islandicus". BMC Molecular Biology. 16: 2. doi:10.1186/s12867-015-0030-z. PMC 4351679. PMID 25880130.
^ Jain SK, Cox MM, Inman RB (August 1994). "On the role of ATP hydrolysis in RecA protein-mediated DNA strand exchange. III. Unidirectional branch migration and extensive hybrid DNA formation". The Journal of Biological Chemistry. 269 (32): 20653–61. PMID 8051165.
^ Ramesh MA, Malik SB, Logsdon JM (January 2005). "A phylogenomic inventory of meiotic genes; evidence for sex in Giardia and an early eukaryotic origin of meiosis". Current Biology. 15 (2): 185–91. doi:10.1016/j.cub.2005.01.003. PMID 15668177.
^ ab Malik SB, Ramesh MA, Hulstrand AM, Logsdon JM (December 2007). "Protist homologs of the meiotic Spo11 gene and topoisomerase VI reveal an evolutionary history of gene duplication and lineage-specific loss". Molecular Biology and Evolution. 24 (12): 2827–41. doi:10.1093/molbev/msm217. PMID 17921483.
^ Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J (2000). "Chapter 8.5: Gene Replacement and Transgenic Animals: DNA Is Transferred into Eukaryotic Cells in Various Ways". Molecular Cell Biology (4th ed.). W. H. Freeman and Company. ISBN 978-0-7167-3136-8.
^ "The Nobel Prize in Physiology or Medicine 2007". The Nobel Foundation. Retrieved December 15, 2008.
^ Drummond DA, Silberg JJ, Meyer MM, Wilke CO, Arnold FH (April 2005). "On the conservative nature of intragenic recombination". Proceedings of the National Academy of Sciences of the United States of America. 102 (15): 5380–5. doi:10.1073/pnas.0500729102. PMC 556249. PMID 15809422.
^ Bloom JD, Silberg JJ, Wilke CO, Drummond DA, Adami C, Arnold FH (January 2005). "Thermodynamic prediction of protein neutrality". Proceedings of the National Academy of Sciences of the United States of America. 102 (3): 606–11. doi:10.1073/pnas.0406744102. PMC 545518. PMID 15644440.
^ ab Carbone MN, Arnold FH (August 2007). "Engineering by homologous recombination: exploring sequence and function within a conserved fold". Current Opinion in Structural Biology. 17 (4): 454–9. doi:10.1016/j.sbi.2007.08.005. PMID 17884462.
^ Otey CR, Landwehr M, Endelman JB, Hiraga K, Bloom JD, Arnold FH (May 2006). "Structure-guided recombination creates an artificial family of cytochromes P450". PLoS Biology. 4 (5): e112. doi:10.1371/journal.pbio.0040112. PMC 1431580. PMID 16594730.
^ Socolich M, Lockless SW, Russ WP, Lee H, Gardner KH, Ranganathan R (September 2005). "Evolutionary information for specifying a protein fold". Nature. 437 (7058): 512–8. doi:10.1038/nature03991. PMID 16177782.
^ Thulasiram HV, Erickson HK, Poulter CD (April 2007). "Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis". Science. 316 (5821): 73–6. doi:10.1126/science.1137786. PMID 17412950.
^ Landwehr M, Carbone M, Otey CR, Li Y, Arnold FH (March 2007). "Diversification of catalytic function in a synthetic family of chimeric cytochrome p450s". Chemistry & Biology. 14 (3): 269–78. doi:10.1016/j.chembiol.2007.01.009. PMC 1991292. PMID 17379142.
^ abc Iglehart JD, Silver DP (July 2009). "Synthetic lethality--a new direction in cancer-drug development". The New England Journal of Medicine. 361 (2): 189–91. doi:10.1056/NEJMe0903044. PMID 19553640.
^ Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (July 2009). "Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers". The New England Journal of Medicine. 361 (2): 123–34. doi:10.1056/NEJMoa0900212. PMID 19553641.
^ Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A (February 2008). "Resistance to therapy caused by intragenic deletion in BRCA2". Nature. 451 (7182): 1111–5. doi:10.1038/nature06548. PMID 18264088.
External links
Library resources about Homologous recombination |
|
| Wikimedia Commons has media related to Homologous recombination. |
Animations – homologous recombination: Animations showing several models of homologous recombination
Homologous recombination: Tempy & Trun: Animation of the bacterial RecBCD pathway of homologous recombination


